Partial Refolding of Ubiquitin:
Molecular Dynamics Simulations of Hydrophobic Collapse.
Alonso and Daggett 1998 Protein Science 7 861-874
Unfolded and partially unfolded conformations of ubiquitin, a globular
protein consisting of 76 residues and a pronounced hydrophobic core,
were simulated under native conditions. The
non-native starting structures were generated during two different thermal
denaturation trajectories. All simulations included all protein and solvent
atoms explicitly, and simulation times ranged from 1-2.4 ns. The seven
non-native starting structures for the simulation under native conditions
had alpha-carbon root mean square (RMS) deviations between 4-11 angstroms
from the crystal structure and radii of gyration as high as 1.3 times that
of the native state. The radii of gyration of the protein calculated
during these seven simulations converged in all but one case with that of a
simulation that started from the crystal structure. In contrast, none of
the structures collapsed when simulated in a 60% methanol:water mixture.
There was a disproportionately high burial of hydrophobic residues
accompanying collapse, but for the most non-native starting structures,
intraprotein hydrogen bonding did not increase concomitant with collapse.
The RMS deviation to the crystal structure decreased from 5 to 3 angstroms
during the course of the simulation of one of the most native-like starting
structures. The less native-like starting structures acquired some native
secondary structure and tertiary contacts during the the simulation but
remained non-native as judged by RMS deviation to the crystal structure.
The least native-like starting structures collapsed, expanded and
collapsed again, and the later collapsed state had and increased burial
of hydrophobic residues relative to the earlier collapsed state.
Although these molecular dynamics simulations are orders of magnitude
too short in duration to follow an entire folding trajectory; taken as
a group representing the changes that occur during simulations of
structures ranging from unfolded to native-like, they suggest that the
the early events in protein folding involve cyclic collapse and
expansion of single protein molecules until approximately native
contacts are formed. Then, there is further consolidation of the core,
better and more spec
hydrogen bonds form.
Native Ubiquitin
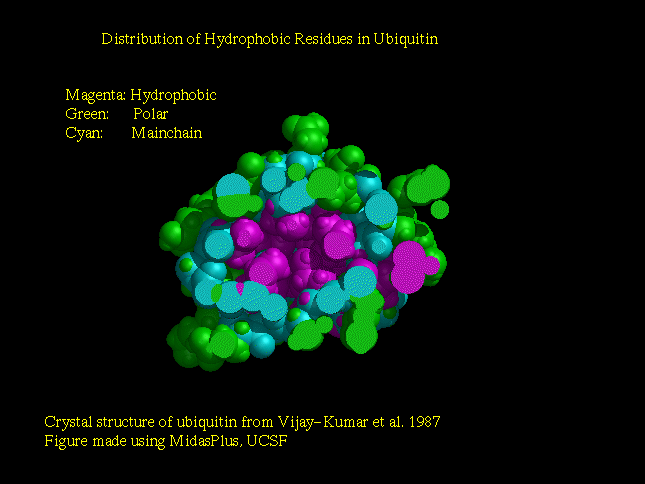
Quicktime Movie of an Initial Collapse
This movie shows the collapse occurring in
first 0.5 ns of a simulation under native conditions
starting with an expanded non-native structure.
This particular starting structure
(I-C in Alonso and Daggett 1998)
had an RMS deviation of 7.6 A from the crystal structure and a radius of
gyration 1.3 times that of the native state.
Only hydrophobic sidechains are displayed.
This movie only covers 0.5 ns, but the simulations lasted 2.4 ns.
The core became better packed in the latter part of the simulation.
MOVIE
Non-Native Starting Structure Becomes more Native-like
I-B in Alonso and Daggett 1998.
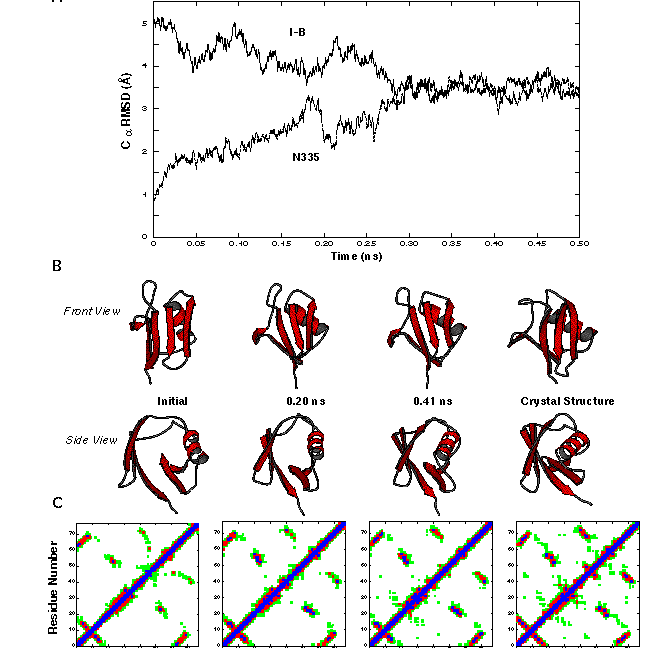
Under native conditons,
structures that were not very different from the native state initially,
relaxed back towards the native state as judged by: RMSD, native contacts,
hydrogen bonds, and secondary structure.
Back to Darwin's Home Page
Back to the Daggett
Group Page